Blast and Fasta Pdf
The first section of videos were created by members of Dr. A profile is built after the initial search that is then used in subsequent searches.

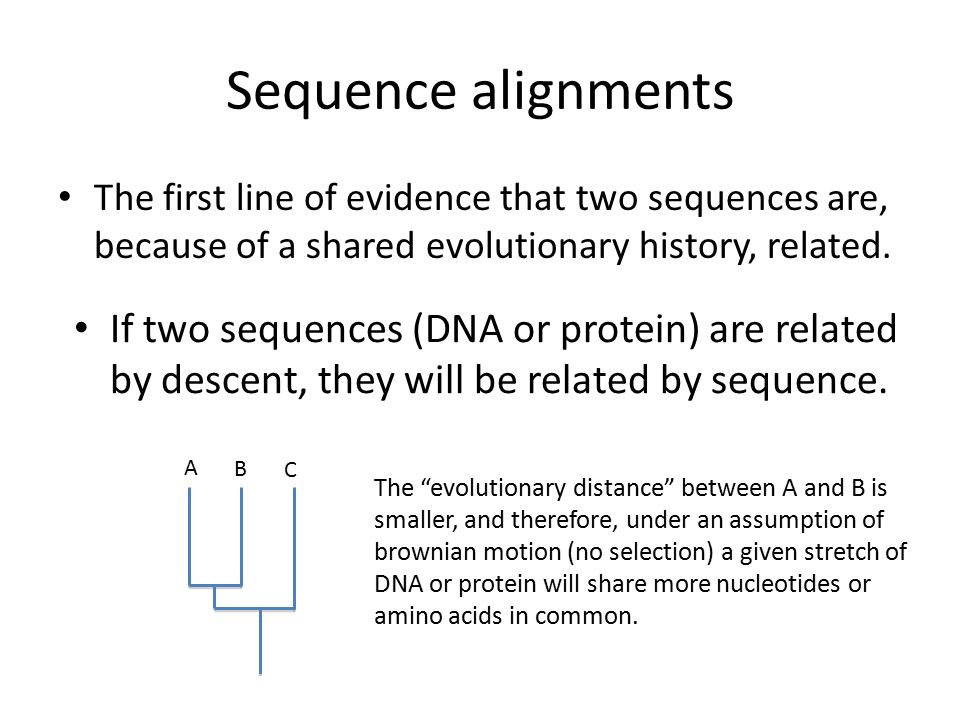
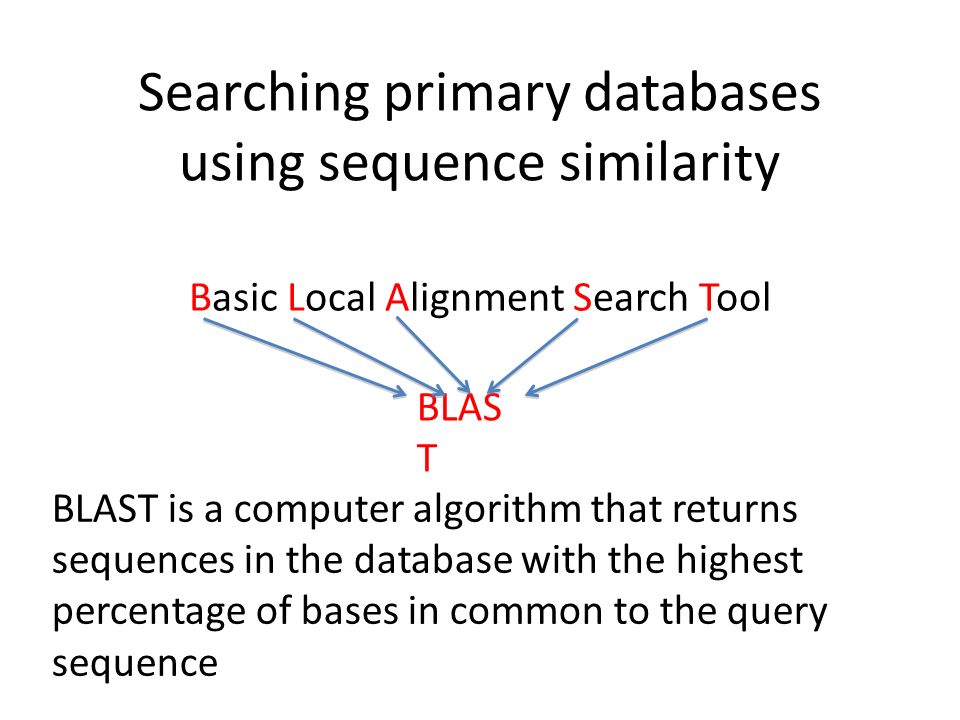
Chapter 11 Assessing Pairwise Sequence Similarity Blast And Fasta Lecture Follows Chapter Pretty Closely This Lecture Is Designed To Introduce You To Ppt Video Online Download
The format has been developed with the advent of large-scale genotyping and DNA sequencing projects such as the 1000 Genomes ProjectExisting formats for genetic data such as General feature format GFF stored all of the genetic data much of.
. It works by finding short stretches of identical or nearly identical letters in two sequences. Job Dispatcher Sequence Analysis Tools Home. A BLAST search enables a researcher to compare a subject protein or nucleotide sequence called a query with a library.
Where available pyani can take advantage of multicore systems and integrates with SGEOGE-type job schedulers for the sequence comparisons. Blast はバイオインフォマティクスにおいてdnaの塩基配列あるいはタンパク質のアミノ酸配列のシーケンスでペアワイズのシーケンスアライメントを行う blast は主にシーケンスの局所的アライメントローカルアライメントを行うために使われる. These are command line programs which run BLAST searches against local downloaded copies of the NCBI BLAST databases or against custom databases formatted for BLAST.
Genome Remapping Service A tool that makes remapping features and annotations simple and straightforward. If you need FASTA for selected sequences from these BLAST databases you can obtain it as follows the sequence of interest is identified by the accession u00001 in this example. Associate membership to the IDM is for up-and-coming researchers fully committed to conducting their research in the IDM who fulfil certain criteria for 3-year terms which are renewable.
Blastdbcmd -entry u00001 -db nr -out. TaqMan probes can also be labeled at the 5-end with the reporter molecule 6- carboxyfluorescein FAM and with a double quencher ZEN. The process may be repeated if desired with new sequences found in each cycle used to refine the profile Altschul et al 1997.
These short strings of characters are called words. Lipman 1997 Gapped BLAST and PSI-BLAST. Use plain FASTA file so seqkit could utilize FASTA index.
The programs will handle either a single large file with multiple FASTA query sequences or you can create a script to send multiple files one at a. In bioinformatics a sequence alignment is a way of arranging the sequences of DNA RNA or protein to identify regions of similarity that may be a consequence of functional structural or evolutionary relationships between the sequences. The Variant Call Format VCF specifies the format of a text file used in bioinformatics for storing gene sequence variations.
Working of FASTA and BLAST. Pyani is a software package and Python3 module that calculates average nucleotide identity ANI and related measures for whole genome comparisons and renders relevant graphical summary output. For this release we have performed a major restructuring of the module that reads the BLAST databases.
The makeblastdb application produces BLAST databases from FASTA files. These utilities run through DOS-like command windows and accept input. In bioinformatics BLAST basic local alignment search tool is an algorithm and program for comparing primary biological sequence information such as the amino-acid sequences of proteins or the nucleotides of DNA andor RNA sequences.
ClinVar A public archive of the relationships between medically important. Position-Specific Iterative BLAST PSI-BLAST is an iterative search using the protein BLAST algorithm. Files UniGene SwissProt Files in the supported formats can be iterated over record by record or indexed and accessed via a Dictionary interface.
Institute leaders such as. It is possible to use completely unstructured or even blank FASTA definition lines but this is not the recommended procedure. In addition to providing BLAST sequence alignment services on the web NCBI also makes these sequence alignment utilities available for download through FTP.
These changes also allow us to support a different threading model threading by query that. The FASTA file format used as input for this software is now largely used by other sequence database search tools such as BLAST and sequence alignment programs Clustal T-Coffee etc. A new generation of protein database search programs.
Sudhir Kumars lab at the Institute for Genomics and Evolutionary Medicine at Temple UniversityThe rest of the videos were produced by users of MEGA. FASTA searches a protein or DNA sequence data bank version 3637b Jun 2015preload9. Assigning a unique identifier to every sequence in the database allows you to retrieve the sequence by identifier and allows you to associate every sequence with a.
Below are links to online video lectures and tutorials for multiple versions of MEGA. Blastdbcmd -entry all -db nr -out nrfsa. The Institute comprises 33 Full and 13 Associate Members with 12 Affiliate Members from departments within the University of Cape Town and 12 Adjunct Members based nationally or internationally.
BLAST to find homologous sequences MAPPING to retrieve GO terms and ANNOTATION to select reliable functions. 1 TaqMan probes are labeled at the 5-end with the reporter molecule 6-carboxyfluorescein FAM and with the quencher Black Hole Quencher 1 BHQ-1 Biosearch Technologies Inc Novato CA at the 3-end. Functional annotation in 3 steps.
Export to PDF Export to Word Pages. The FASTA package is available from the University of Virginia and the European Bioinformatics Institute. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrixGaps are inserted between the.
FASTA and BLAST are the software tools used in bioinformatics. Pairwise sequence alignment methods such as BLAST and FASTA use position-independent subsitution score matrices such as BLOSUM and PAM but the desirability of position-specific models was recog-nized even before BLAST and FASTA were written3 Several groups 3 R. Both from standalone and WWW Blast Clustalw FASTA GenBank PubMed and Medline ExPASy files like Enzyme and Prosite SCOP including dom.
Webb Miller and David J. If you need FASTA from these BLAST databases you can obtain it as follows. First 12 bases zcat hairpinfagz seqkit subseq -r 112 Last 12 bases zcat hairpinfagz seqkit subseq -r -12-1 Subsequences without first and last 12 bases zcat hairpinfagz seqkit subseq -r 13-13 Get subsequence by GTF file.
For multithreaded searches these changes reduce the number of mutex calls result in the use of fewer file pointers and reduce the number of calls to memory map. This allows BLAST searches to be performed on local platforms against databases downloaded from NCBI or created locally. Similar amino acid se-quences.
The European Bioinformatics Institute EMBL-EBI is an Intergovernmental Organization IGO which as part of the European Molecular Biology Laboratory EMBL family focuses on research and services in bioinformaticsIt is located on the Wellcome Genome Campus in Hinxton near Cambridge and employs over 600 full-time equivalent FTE staff. PDF reports custom fasta annot and generic data formats X BioMart data import Verteb Metazoa Plants Protists Fungi X. 最初のバージョンは fastp という名前.
Chance or common ancestry. Both BLAST and FASTA use a heuristic word method for fast pairwise sequence alignment. Fact Sheets to Download PDF Genome Reference Consortium GRC Ensuring that the reference assemblies continue to grow as our understanding of these genomes evolve.

Blast

Fasta
Chapter 11 Assessing Pairwise Sequence Similarity Blast And Fasta Lecture Follows Chapter Pretty Closely This Lecture Is Designed To Introduce You To Ppt Video Online Download
Chapter 11 Assessing Pairwise Sequence Similarity Blast And Fasta Lecture Follows Chapter Pretty Closely This Lecture Is Designed To Introduce You To Ppt Video Online Download

Comments
Post a Comment